近年來,隨著檢驗技術的日新月異,人類基因體學研究與分子檢測技術迅速發展,許多新醫學檢驗項目從實驗室中被研發出來至臨床應用,實驗室自行研發檢驗技術(Laboratory Developed Tests,LDTs)成為快速成長的服務項目,更被視為實現預防醫學與精準醫療的重要推手。而隨著LDTs的大幅增加及商業化發展,如何在創新發展與監管之間拿捏,亦成為各國監理機關的重要課題。
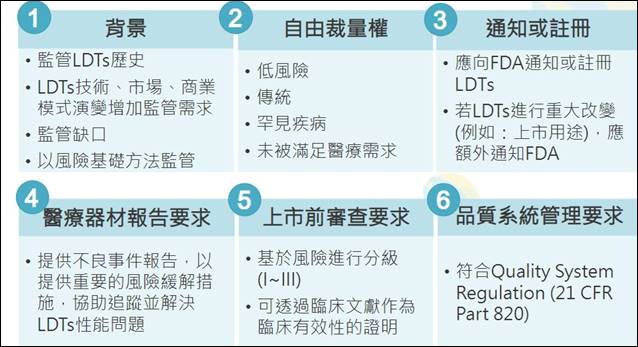
LDTs在美國是由聯邦醫療保險和補助服務中心(The Centers for Medicare and Medicaid Services,CMS)進行實驗室認證,隨著LDTs的數量及類型大幅增加、發展越趨商業化,美國食品藥物管理局(Food and Drug Administration,FDA) 在2014年發布LDTs監管架構指引草案,以風險基礎方法對LDTs加以監管和進行分級。除了應向FDA通知或註冊LDTs,若進行重大改變也必須額外通知FDA,結果引發各界抨擊。FDA另於2017年發布LDTs討論文件,尚未執行此監管架構。
2018年,美國國會議員提出由FDA監管的《評估精準且先進的體外臨床檢驗技術發展法》(Verifying
Accurate,Leading-edge /VCT Development Act, VALID Act)草案、參議院議員另提出由CMS監管之《評估美國驗室之創新檢測法》(Verified Innovative Testing in American Laboratories Act,VITAL Act)草案,雖然在116屆和117屆美國國會都曾提出過法案,至今皆仍未通過。
VALID Act將LDTs列為「體外臨床檢測」(In vitro clinical tests,IVCTs),認為應有獨立的監管架構,必須依照對病人和公眾危害嚴重程度,分為高、中、低風險。除了要求臨床確效報告使用真實世界數據,還要制定FDA諮詢機制並舉行公聽會。呂政諺指出,VALID Act比《臨床實驗室改進修正案》(CLIA)更為嚴格,相關規定涵蓋管理職責、品質稽核要求、人員、設計管理、文件管理等各方面要求,反對者認為VALID Act是在阻礙創新,加上FDA缺足夠的資源履行這些義務,不可預測的監管流程將會造成成本增加。2020年美國衛生及公共服務部(United States Department of Health and Human Services,HHS)也禁止FDA針對LDTs進行上市前審查。不過,無論VALID Act是否通過,FDA仍可能以其他方式監管LDTs。
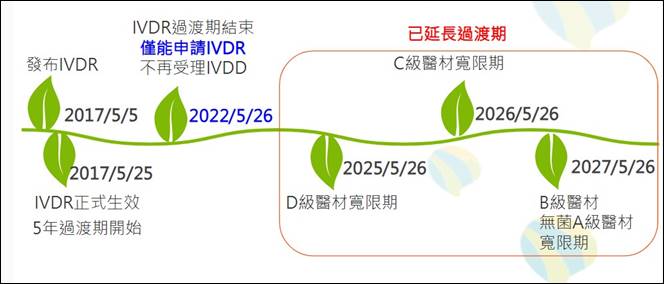
IVDR將產品依風險分成A~D級,需第三機構驗證的產品從20%大幅提高至80%。原先取得IVDD驗證的產品,仍須透過第三方機構進行IVDR驗證,不僅技術文件及臨床數據要求增加,並強化產品可追溯性,要求透過歐盟醫療器材資料庫(EU medical devices database,EUDAMED)向大眾公開安全與性能摘要。此外,除了上市後監督,每五年還會對產品進行一次無預警稽核。考量到目前第三方機構驗證能量不足,歐盟已依醫材風險等級不同提供延長過渡期。
日本則未設立LDTs專法,將LDTs稱為「自家調整檢查法」,醫療用途的基因檢測僅能在醫療機構中執行。2017年,日本修正及《醫療法》及《臨床檢驗師法》,依檢驗行為及檢驗場所分為醫療機構自行檢查、委託其他醫療機構檢查、委託醫事檢查所檢查,主管機關為厚生勞動省(Ministry of Health, Labour and Welfare,MHLW)和醫藥品醫療器材綜合機構(Pharmaceuticals and Medical Devices Agency,PMDA)。日本目前已核准3項用於癌症基因的次世代基因定序(NGS)檢測上市,由國民健康保險部分負擔,只能在獲得MHLW核可的醫院下進行檢測。