人類在17世紀末就已經開始使用疫苗及各種其他生物製品,包含病毒、治療血清、毒素和抗毒素。1901年秋天,美國聖路易市有兒童因接種了白喉抗毒素而死於破傷風,而紐澤西州的卡姆登也有兒童接種了天花疫苗後死於破傷風[2],這兩起事件幾乎在同一時間發生。在這兩種生物製劑被破傷風汙染並導致兒童死亡的事件之後,美國國會於1902年即通過了 《生物製劑控制法(Biologics Control Act)》,主管機關為國家衛生研究院 (National Institutes of Health, NIH)的前身。此時,聯邦政府並沒有對非生物藥物進行管制[3]。
1906年的《食品及藥物法(Food and Drugs Act)》的主管機關為農業部化學局(Bureau of Chemistry, Department of Agriculture)[4],目的在禁止已上市藥品的摻假 (adulteration) 及濫標品牌 (misbranding)[5]。到了1937年,S. E. Massengill,一間生產高品質藥品的公司,將一種抗菌劑磺胺 (sulfanilamide) 溶於二甘醇 (diethylene glycol) 製成液體製劑,但卻沒有做動物毒性測試,Massengill也沒有預先檢查藥物的安全性,而當時FDA也沒有進行這種測試的慣例。這個名為磺胺酏劑 (Elixir Sulfanilamide) 的藥品最終造成107人死亡 (其中許多是兒童) [6]。這個事件促使當時美國總統羅斯福簽署通過了1938年的《聯邦食品、藥物及化妝品法 (Federal Food, Drug, and Cosmetic Act, FDCA)》,規定所有新藥上市前必須通過安全性的審查[7]。到了1944年,美國國會修訂生物製劑控制法並將之編到《公共衛生服務法 (Public Health Service Act, PHSA)》第351節[8]。
橘皮書和專利連結制度的誕生由於測試新藥的安全性和有效性要求製造商對人類受試者進行昂貴且耗時的測試,新藥申請程序導致藥品價格急劇上漲。增加測試要求同樣導致在新藥申請的提交與其許可之間延遲超過3年[12]。然而,學名藥製造商想要上市1962年以後原廠藥的學名藥也須要進行臨床調查,在成本增加的情況底下,這阻礙了學名藥廠上市低價學名藥[13]。為了衡平創新工業、學名藥工業及消費者三方的利益,美國在1984年9月頒布了《藥價競爭及專利期間回復法案 (Drug Price Competition and Patent Term Restoration Act)》,一般又稱為哈奇-韋克斯曼法案 (Hatch-Waxman Act),包含了四個部分:(1)建立了學名藥簡易許可途徑:簡易新藥申請(Abbreviated New Drug Application, ANDA);(2)裁決學名藥製造商挑戰原廠藥製造商的市場專屬權的系統;(3)確保原廠藥許可的無競爭期間;(4)延長品牌藥的市場專屬權[14]。
FDA建立了證明療效相等性 (Therapeutic Equivalence) 的方式,只要學名藥與原廠藥具有藥學相等性(pharmaceutical equivalence)及生體相等性(bioequivalence),即可認定學名藥與原廠藥是療效相等的而可替代(substitution)(圖1)。藥學相等性必須符合三個標準:(1)含有相同的有效成分;(2)具有相同的劑型和給藥途徑;(3)具有相同的單位含量(strength)或濃度。生體相等性測量可由達到藥物最大血清(或血漿)濃度的時間(Cmax),或由血清濃度隨時間變化所定義的曲線下面積(AUC)來描繪。FDA將生體相等性定義為,原廠藥對學名藥的AUC及Cmax之比的90%信賴區間落在可接受區間0.80-1.25內(稱為「-20%/+25%規則」)(如圖1所示)。如果學名藥製造商可以證明藥學相等性及生體相等性,則不需要額外的二期及三期臨床試驗[15]。FDA將許可的藥品列在「具療效相等性評估之許可藥品目錄(Approved Drug Products with Therapeutic Equivalence Evaluations)」,稱為橘皮書 (Orange Book),一本FDA已許可藥品有替代學名藥的概要。橘皮書讓健保決策者輕鬆決定哪些學名藥與表列的參考品牌藥具有生體相等性及藥學相等性。
然而,2006年美國最高法院在eBay Inc. v. MercExchange, L.L.C.案中闡明,法院判斷永久禁制令核發與否時,必須考量4個因素,原告必須證明:(1)原告遭受不可回復之損害;(2)法律上提供救濟之方式不足以彌補該損害;(3)在衡量原告與被告雙方損益後,衡平救濟是正當的;(4)核發永久禁制令不會損及公眾利益。2009年的生物製劑價格競爭與創新法案在2007年進行關鍵協商時受到eBay案很大的影響,最終並沒有「自動啟動暫停核發學名藥許可」的平行規定。
該法規定在新分子實體 (new molecular entities, NMEs) 許可後的5年內不得提交學名藥申請[25],這保證了任何製造商即使沒有專利保護,也至少有5年時間可賺取收入以回收研發成本並獲得獨占的利潤[26]。原廠藥製造商認為1962年的Kefauver-Harris Amendment要求冗長的臨床調查及FDA審查不公平的縮短了他們的有效專屬權期限。因為專利核准時可能早於FDA許可,當FDA許可藥品上市時,可能只剩很少或根本沒有專利期間。Hatch-Waxman Act以「專利期間回復」來解決這個議題,在原來的專利期間增加額外的時間來補償因臨床調查及FDA審查所失去的專利期間[27]。
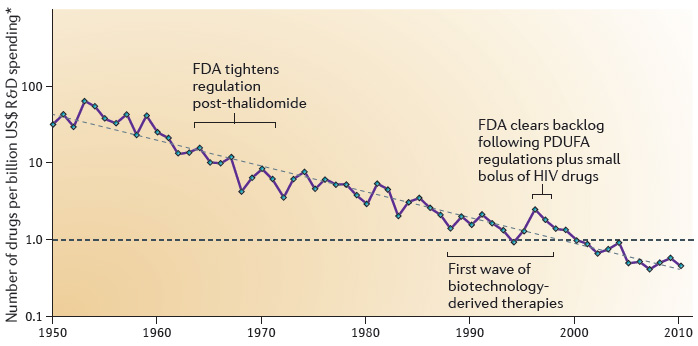
過去60年來,隨著科學、技術及管理的進展,藥物研發的效率應該要提高。然而,自1950年以來,每10億美元研發費用所許可的新藥數量大約每九年減少一半(圖4),Sanford C. Bernstein Limited的Jack W. Scannell把這個現象稱為「Eroom’s Law」,因為這與積體電路的Moore’s Law相反[41]。自2000年開始,每個許可的新藥平均花費的研發費用超過10億美金。
圖4:藥品研發的Eroom’s Law 資料來源:Jack W. Scannell et al., Diagnosing the Decline in Pharmaceutical R&D Efficiency, NATURE REVIEWS: DRUG DISCOVERY 11 (2012), p. 191-192
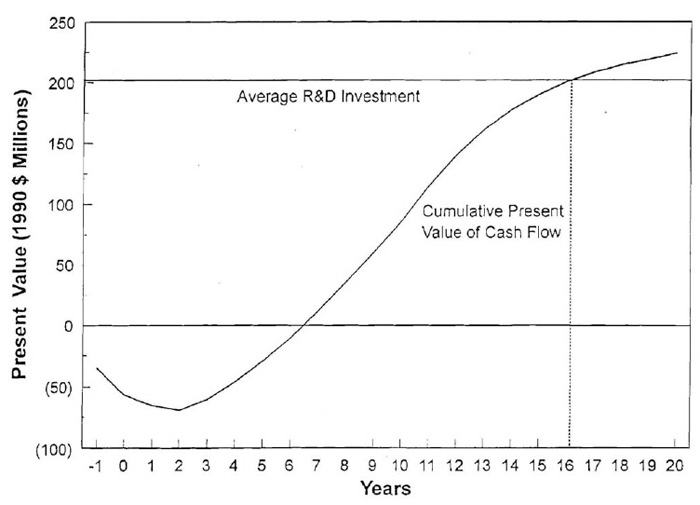
圖5:1980年至1984年間導入之化學新藥的現金流與研發投資的累積現值[42]
資料來源:Henry Grabowski, Data Exclusivity for New Biological Entities,
(Duke University Department of Economics Working Paper No. 2., June 2007), p. 34
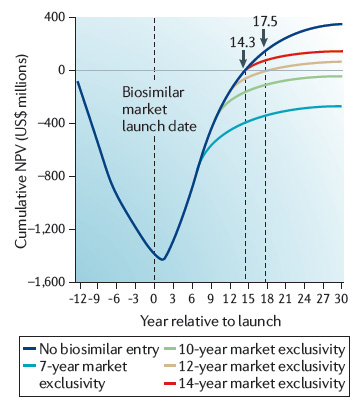
圖6:各種上市情景的代表性生物製劑現金流的累計淨現值
資料來源:Henry Grabowski, Data exclusivity for biologics,
NATURE REVIEWS: DRUG DISCOVERY 10 (2011), p. 15
Ronald Hamowy, Medical Disasters and the Growth of the FDA, Independent Policy Report (2010), p. 3-4
Krista Hessler Carver et al., An Unofficial Legislative History of the Biologics Price Competition and Innovation Act of 2009, FOOD AND DRUG LAW JOURNAL 65 (2010), p. 682
1930年更名為食品藥物管理局(Food and Drug Administration, FDA)
同註2, p. 672
同註1, p. 5-6
同註2,p. 673
同前註,p. 682-683
李尚仁,沙利竇邁藥害事件的兩位醫學英雄,科學發展536(2017.8),頁78-80
同註2,p. 674
Aaron S. Kesselheim et al., Hatch-Waxman Turns 30: Do We Need a Re-Designed Approach for the Modern Era?, YALE JOURNAL OF HEALTH POLICY, LAW, AND ETHICS 15:2 (2015), pp. 297
Joel Graham, The Legality of Hatch-Waxman Pharmaceutical Settlements: Is the Terazosin Test the Proper Prescription?, WASHINGTON UNIVERSITY LAW REVIEW 84 (2006), pp. 432-433
同註11,p. 297
同註11,p. 301
同註11,p. 302
21 U.S.C. §355(b)(1)
21 U.S.C. §355(j)(2)(B)
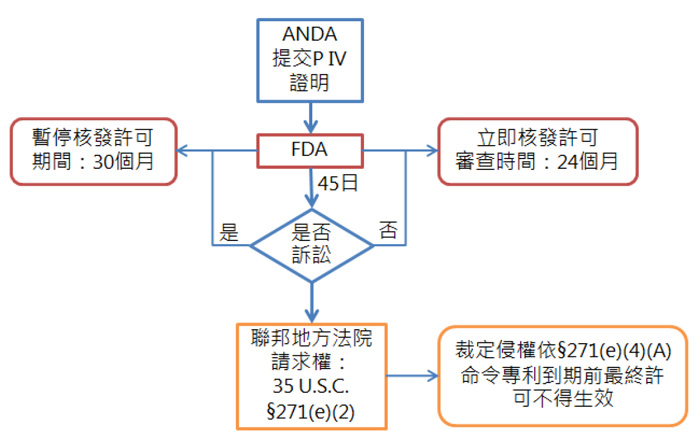
35 U.S.C. §271(e)(2)
21 U.S.C. §355(j)(5)(B)(iii)
同註2,p. 697
同註18
35 U.S.C. §271(e)(4)(A)
同註18
21 U.S.C. §355(j)(5)(B)(iv)
21 U.S.C. §355(j)(5)(F)(ii)
同註11,p. 305
同註11, p.306
傑瑞米‧葛林(Jeremy A. Greene),便宜沒好藥?一段學名藥和當代醫療的糾葛 (Generic-The Unbranding of Modern Medicine),左岸文化,頁193-194
同註28,頁182
同註28,頁195-196、201-209
同註11,p. 307
同註11,p. 310
同註2,p. 688-695
同註2,p. 706-707
同註2,p. 716-723
同註2,p. 739-746
同註2,p. 746-761
同註2,p. 806-816
Jack W. Scannell et al., Diagnosing the Decline in Pharmaceutical R&D Efficiency, NATURE REVIEWS: DRUG DISCOVERY 11 (2012), p. 191-192
Henry Grabowski, Data Exclusivity for New Biological Entities, (Duke University Department of Economics Working Paper No. 2., June 2007), p. 34